Advanced Renal Cell Carcinoma
KEYTRUDA, in combination with LENVIMA, is indicated for the first-line treatment of adult patients with advanced renal cell carcinoma (RCC).
KEYTRUDA
Immune-mediated adverse reactions, which may be severe or fatal, can occur in any organ system or tissue, can affect more than one body system simultaneously, and can occur at any time after starting treatment or after discontinuation of treatment. Important immune‑mediated adverse reactions listed here may not include all possible severe and fatal immune‑-mediated adverse reactions.

Monitor patients closely for symptoms and signs that may be clinical manifestations of underlying immune-mediated adverse reactions.

Withhold or permanently discontinue KEYTRUDA depending on severity of the immune-mediated adverse reaction.

Patient Counseling Information
 Immune-Mediated Pneumonitis
Immune-Mediated Pneumonitis[see Warnings and Precautions]
Grade 2
Withhold KEYTRUDAa
Grade 3 or 4
Permanently discontinue KEYTRUDA
aResume in patients with complete or partial resolution (Grade 0 to 1) after corticosteroid taper. Permanently discontinue if no complete or partial resolution within 12 weeks of initiating steroids or inability to reduce prednisone to 10 mg per day or less (or equivalent) within 12 weeks of initiating steroids.
Immune-Mediated Colitis[see Warnings and Precautions]
Grade 2 or 3
Withhold KEYTRUDAa
Grade 4
Permanently discontinue KEYTRUDA
aResume in patients with complete or partial resolution (Grade 0 to 1) after corticosteroid taper. Permanently discontinue if no complete or partial resolution within 12 weeks of initiating steroids or inability to reduce prednisone to 10 mg per day or less (or equivalent) within 12 weeks of initiating steroids.
Immune-Mediated Hepatitis With No Tumor Involvement of the Liver[see Warnings and Precautions]
AST or ALT increases to more than 3 and up to 8 times ULN
Withhold KEYTRUDAa
Total bilirubin increases to more than 1.5 and up to 3 times ULN
Withhold KEYTRUDAa
AST or ALT increases to more than 8 times ULN
Permanently discontinue KEYTRUDA
Total bilirubin increases to more than 3 times ULN
Permanently discontinue KEYTRUDA
aResume in patients with complete or partial resolution (Grade 0 to 1) after corticosteroid taper. Permanently discontinue if no complete or partial resolution within 12 weeks of initiating steroids or inability to reduce prednisone to 10 mg per day or less (or equivalent) within 12 weeks of initiating steroids.
Immune-Mediated Hepatitis With Tumor Involvement of the Liver[see Warnings and Precautions]
Baseline AST or ALT is more than 1 and up to 3 times ULN and increases to more than 5 and up to 10 times ULN
Withhold KEYTRUDAb
Baseline AST or ALT is more than 3 times and up to 5 times ULN and increases to more than 8 times and up to 10 times ULN
Withhold KEYTRUDAb
Baseline AST or ALT increases to more than 10 times ULN
Permanently discontinue KEYTRUDA
Total bilirubin increases to more than 3 times ULN
Permanently discontinue KEYTRUDA
aIf AST and ALT are less than or equal to ULN at baseline, withhold or permanently discontinue KEYTRUDA based on recommendations for hepatitis with no liver involvement.
bResume in patients with complete or partial resolution (Grade 0 to 1) after corticosteroid taper. Permanently discontinue if no complete or partial resolution within 12 weeks of initiating steroids or inability to reduce prednisone to 10 mg per day or less (or equivalent) within 12 weeks of initiating steroids.
Adrenal Insufficiency (Immune-Mediated Endocrinopathies)[see Warnings and Precautions]
Grade 2 or higher
Initiate symptomatic treatment, including hormone replacement as clinically indicated. Withhold KEYTRUDA depending on severity
Grade 3 or 4
Initiate symptomatic treatment, including hormone replacement as clinically indicated. Withhold until clinically stable or permanently discontinue KEYTRUDA depending on severity
Hypophysitis (Immune-Mediated Endocrinopathies)[see Warnings and Precautions]
Grade 3 or 4
Withhold until clinically stable or permanently discontinue KEYTRUDA depending on severity
Thyroid Disorders (Immune-Mediated Endocrinopathies)[see Warnings and Precautions]
Grade 3 or 4
Withhold until clinically stable or permanently discontinue KEYTRUDA depending on severity
Type 1 Diabetes Mellitus, Which Can Present With Diabetic Ketoacidosis (Immune-Mediated Endocrinopathies)[see Warnings and Precautions]
Grade 3 or 4
Withhold until clinically stable or permanently discontinue KEYTRUDA depending on severity
Immune-Mediated Nephritis With Renal Dysfunction[see Warnings and Precautions]
Grade 2 or 3 increased blood creatinine
Withhold KEYTRUDAa
Grade 4 increased blood creatinine
Permanently discontinue KEYTRUDA
aResume in patients with complete or partial resolution (Grade 0 to 1) after corticosteroid taper. Permanently discontinue if no complete or partial resolution within 12 weeks of initiating steroids or inability to reduce prednisone to 10 mg per day or less (or equivalent) within 12 weeks of initiating steroids.
Immune-Mediated Dermatologic Adverse Reactions[see Warnings and Precautions]
Exfoliative Dermatologic Conditions: Suspected SJS, TEN, or DRESS
Withhold KEYTRUDAa
Exfoliative Dermatologic Conditions: Confirmed SJS, TEN, or DRESS
Permanently discontinue KEYTRUDA
aResume in patients with complete or partial resolution (Grade 0 to 1) after corticosteroid taper. Permanently discontinue if no complete or partial resolution within 12 weeks of initiating steroids or inability to reduce prednisone to 10 mg per day or less (or equivalent) within 12 weeks of initiating steroids.
Immune-Mediated Myocarditis[see Warnings and Precautions]
Grade 2, 3, or 4
Permanently discontinue KEYTRUDA
Immune-Mediated Neurological Toxicities[see Warnings and Precautions]
Grade 2
Withhold KEYTRUDAa
Grade 3 or 4
Permanently discontinue KEYTRUDA
aResume in patients with complete or partial resolution (Grade 0 to 1) after corticosteroid taper. Permanently discontinue if no complete or partial resolution within 12 weeks of initiating steroids or inability to reduce prednisone to 10 mg per day or less (or equivalent) within 12 weeks of initiating steroids.
Complications of Allogeneic Hematopoietic Stem Cell Transplantation (HSCT)[see Warnings and Precautions]
Increased Mortality in Patients With Multiple Myeloma When KEYTRUDA Is Added to a Thalidomide Analogue and Dexamethasone[see Warnings and Precautions]
Other Immune-Mediated Adverse Reactions (IMARs)[see Warnings and Precautions]
Infusion-Related Reactions[see Warnings and Precautions]
Grade 1 or 2
Interrupt or slow the rate of KEYTRUDA infusion
Grade 3 or 4
Stop infusion and permanently discontinue KEYTRUDA
Embryo-Fetal Toxicity[see Warnings and Precautions]
Lactation (Use in Specific Populations)LENVIMA
Withhold, reduce, or discontinue LENVIMA based on the type and/or severity (grade) of the adverse reaction.
Recommended dosage reductions for LENVIMA for patients with advanced renal cell carcinoma or advanced endometrial carcinomaa

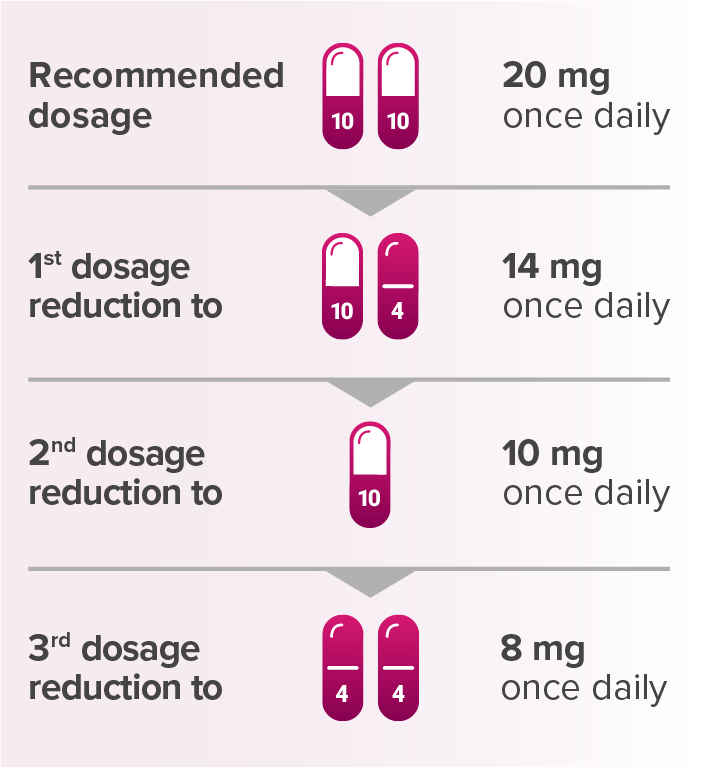
aWhen administered with KEYTRUDA.
Capsules are not shown at actual size.
Hypertension[see Warnings and Precautions]


Grade 3
Withhold LENVIMA for Grade 3 that persists despite optimal antihypertensive therapy
Resume LENVIMA at a reduced dose when hypertension is controlled at ≤ Grade 2
Grade 4
Permanently discontinue LENVIMA
 Cardiac Dysfunction
Cardiac Dysfunction[see Warnings and Precautions]
Grade 3
Withhold LENVIMA until improves to Grade 0 to 1 or baseline
Resume at a reduced dose or discontinue LENVIMA depending on the severity and persistence of adverse reaction
Grade 4
Permanently discontinue LENVIMA
Arterial Thromboembolic Events[see Warnings and Precautions]
Any grade of arterial thromboembolic event
Permanently discontinue LENVIMA
Hepatotoxicity[see Warnings and Precautions]
Grade 3 or 4
Withhold LENVIMA until improves to Grade 0 to 1 or baseline
Either resume at a reduced dose or discontinue LENVIMA depending on severity and persistence of hepatotoxicity
Permanently discontinue LENVIMA for hepatic failure
Renal Failure or Impairment[see Warnings and Precautions]
Grade 3 or 4
Withhold LENVIMA until improves to Grade 0 to 1 or baseline
Resume at a reduced dose or discontinue LENVIMA depending on severity and persistence of renal impairment
Proteinuria[see Warnings and Precautions]
≥ 2 g proteinuria in 24 hours
Withhold LENVIMA until ≤ 2 g of proteinuria per 24 hours
Resume LENVIMA at a reduced dose
Permanently discontinue LENVIMA for nephrotic syndrome
Diarrhea[see Warnings and Precautions]
Grade 2 or 3; Persistent or intolerable adverse reaction:
Withhold LENVIMA until improves to Grade 0 to 1 or baseline
Resume LENVIMA at a reduced dose
Grade 4
Permanently discontinue LENVIMA
Gastrointestinal Perforation[see Warnings and Precautions]
Any grade of gastrointestinal perforation
Permanently discontinue LENVIMA
Fistula Formation[see Warnings and Precautions]
Grade 3 or 4
Permanently discontinue LENVIMA
QT Interval Prolongation[see Warnings and Precautions]
For QT interval > 500 ms or for > 60 ms increase in baseline QT interval
Withhold LENVIMA until improves to ≤ 480 ms or baseline
Resume LENVIMA at a reduced dose
Hypocalcemia[see Warnings and Precautions]
Grade 2 or 3; Persistent or intolerable adverse reaction:
Withhold LENVIMA until improves to Grade 0 to 1 or baseline
Resume LENVIMA at a reduced dose
Grade 4
Withhold LENVIMA until improves to Grade 0 to 1 or baseline
Resume LENVIMA at a reduced dose
Permanently discontinue LENVIMA depending on severity
Reversible Posterior Leukoencephalopathy Syndrome (RPLS)[see Warnings and Precautions]
Any grade of reversible posterior leukoencephalopathy syndrome
Withhold and resume at a reduced dose upon recovery or permanently discontinue LENVIMA depending on severity and persistence of neurologic symptoms
Hemorrhagic Events[see Warnings and Precautions]
Grade 2 or 3; Persistent or intolerable adverse reaction:
Withhold LENVIMA until improves to Grade 0 to 1 or baseline
Resume LENVIMA at a reduced dose
Grade 4
Permanently discontinue LENVIMA
Impairment of Thyroid-Stimulating Hormone Suppression/Thyroid Dysfunction[see Warnings and Precautions]
Impaired Wound Healing[see Warnings and Precautions]
Withhold LENVIMA for at least 1 week prior to elective surgery
Do not administer LENVIMA for at least 2 weeks following major surgery and until adequate wound healing
Osteonecrosis of the Jaw (ONJ)[see Warnings and Precautions]
Withhold LENVIMA for at least 1 week prior to scheduled dental surgery or invasive dental procedures, if possible
Withhold LENVIMA if ONJ develops and restart based on clinical judgment of adequate resolution
Other Adverse Reactions[See Warnings and Precautions for Diarrhea, Hypocalcemia, and Hemorrhagic Events]
Grade 2 or 3; Persistent or intolerable adverse reaction; or Grade 4 laboratory abnormalities
Withhold LENVIMA until improves to Grade 0 to 1 or baseline
Resume LENVIMA at reduced dose
Grade 4 adverse reaction
Permanently discontinue LENVIMA
Embryo-Fetal Toxicity[see Warnings and Precautions]
Lactation (Use in Specific Populations)DEFINITIONS AND TERMS
Common Terminology Criteria for Adverse Events (CTCAE) grading definitions are listed according to version 4.0, which is the version that is used in the Prescribing Information for KEYTRUDA and for LENVIMA.
aA semicolon indicates “or” within the description of the grade.
Instrumental ADL refer to preparing meals, shopping for groceries or clothes, using the telephone, managing money, etc.2
Self-care ADL refer to bathing, dressing and undressing, feeding self, using the toilet, taking medications, and not bedridden.2
Advanced Renal Cell Carcinoma
KEYTRUDA, in combination with LENVIMA, is indicated for the first-line treatment of adult patients with advanced renal cell carcinoma (RCC).
Advanced Endometrial Carcinoma
KEYTRUDA, in combination with LENVIMA, is indicated for the treatment of adult patients with advanced endometrial carcinoma that is mismatch repair proficient (pMMR) or not microsatellite instability-high (MSI-H) as determined by an FDA-approved test, who have disease progression following prior systemic therapy in any setting and are not candidates for curative surgery or radiation.
Severe and Fatal Immune-Mediated Adverse Reactions
Immune-Mediated Pneumonitis
Immune-Mediated Colitis
Hepatotoxicity and Immune-Mediated Hepatitis
KEYTRUDA as a Single Agent
Immune-Mediated Endocrinopathies
Adrenal Insufficiency
Hypophysitis
Thyroid Disorders
Type 1 Diabetes Mellitus (DM), Which Can Present With Diabetic Ketoacidosis
Immune-Mediated Nephritis With Renal Dysfunction
Immune-Mediated Dermatologic Adverse Reactions
Other Immune-Mediated Adverse Reactions
Infusion-Related Reactions
Complications of Allogeneic Hematopoietic Stem Cell Transplantation (HSCT)
Increased Mortality in Patients With Multiple Myeloma
Embryofetal Toxicity
Adverse Reactions
Lactation
Hypertension
Cardiac Dysfunction
Arterial Thromboembolic Events
Hepatotoxicity
Renal Failure or Impairment
Proteinuria
Diarrhea
Fistula Formation and Gastrointestinal Perforation
QT Interval Prolongation
Hypocalcemia
Reversible Posterior Leukoencephalopathy Syndrome (RPLS)
Hemorrhagic Events
Impairment of Thyroid Stimulating Hormone Suppression/Thyroid Dysfunction
Impaired Wound Healing
Osteonecrosis of the Jaw (ONJ)
Embryo-Fetal Toxicity
Adverse Reactions
Use in Specific Populations

KEYTRUDA, in combination with LENVIMA, is indicated for the first-line treatment of adult patients with advanced renal cell carcinoma (RCC).
KEYTRUDA, in combination with LENVIMA, is indicated for the treatment of adult patients with advanced endometrial carcinoma that is mismatch repair proficient (pMMR) or not microsatellite instability-high (MSI-H) as determined by an FDA-approved test, who have disease progression following prior systemic therapy in any setting and are not candidates for curative surgery or radiation.
Advanced Renal Cell Carcinoma
KEYTRUDA, in combination with LENVIMA, is indicated for the first-line treatment of adult patients with advanced renal cell carcinoma (RCC).
Advanced Endometrial Carcinoma
KEYTRUDA, in combination with LENVIMA, is indicated for the treatment of adult patients with advanced endometrial carcinoma that is mismatch repair proficient (pMMR) or not microsatellite instability-high (MSI-H) as determined by an FDA-approved test, who have disease progression following prior systemic therapy in any setting and are not candidates for curative surgery or radiation.
Severe and Fatal Immune-Mediated Adverse Reactions: KEYTRUDA is a monoclonal antibody that belongs to a class of drugs that bind to either the programmed death receptor-1 (PD-1) or the programmed death ligand 1 (PD-L1), blocking the PD-1/PD-L1 pathway, thereby removing inhibition of the immune response, potentially breaking peripheral tolerance and inducing immune-mediated adverse reactions. Immune-mediated adverse reactions, which may be severe or fatal, can occur in any organ system or tissue, can affect more than one body system simultaneously, and can occur at any time after starting treatment or after discontinuation of treatment. Important immune-mediated adverse reactions listed here may not include all possible severe and fatal immune-mediated adverse reactions.
Hypertension: In differentiated thyroid cancer (DTC), hypertension occurred in 73% of patients on LENVIMA (44% grade 3-4). In advanced renal cell carcinoma (RCC), hypertension occurred in 42% of patients on LENVIMA + everolimus (13% grade 3). Systolic blood pressure ≥160 mmHg occurred in 29% of patients, and 21% had diastolic blood pressure ≥100 mmHg. In unresectable hepatocellular carcinoma (HCC), hypertension occurred in 45% of LENVIMA-treated patients (24% grade 3). Grade 4 hypertension was not reported in HCC.
Severe and Fatal Immune-Mediated Adverse Reactions
Immune-Mediated Pneumonitis
Immune-Mediated Colitis
Hepatotoxicity and Immune-Mediated Hepatitis
KEYTRUDA as a Single Agent
Immune-Mediated Endocrinopathies
Adrenal Insufficiency
Hypophysitis
Thyroid Disorders
Type 1 Diabetes Mellitus (DM), Which Can Present With Diabetic Ketoacidosis
Immune-Mediated Nephritis With Renal Dysfunction
Immune-Mediated Dermatologic Adverse Reactions
Other Immune-Mediated Adverse Reactions
Infusion-Related Reactions
Complications of Allogeneic Hematopoietic Stem Cell Transplantation (HSCT)
Increased Mortality in Patients With Multiple Myeloma
Embryofetal Toxicity
Adverse Reactions
Lactation
Hypertension
Cardiac Dysfunction
Arterial Thromboembolic Events
Hepatotoxicity
Renal Failure or Impairment
Proteinuria
Diarrhea
Fistula Formation and Gastrointestinal Perforation
QT Interval Prolongation
Hypocalcemia
Reversible Posterior Leukoencephalopathy Syndrome (RPLS)
Hemorrhagic Events
Impairment of Thyroid Stimulating Hormone Suppression/Thyroid Dysfunction
Impaired Wound Healing
Osteonecrosis of the Jaw (ONJ)
Embryo-Fetal Toxicity
Adverse Reactions
Use in Specific Populations
Severe and Fatal Immune-Mediated Adverse Reactions: KEYTRUDA is a monoclonal antibody that belongs to a class of drugs that bind to either the programmed death receptor-1 (PD-1) or the programmed death ligand 1 (PD-L1), blocking the PD-1/PD-L1 pathway, thereby removing inhibition of the immune response, potentially breaking peripheral tolerance and inducing immune-mediated adverse reactions. Immune-mediated adverse reactions, which may be severe or fatal, can occur in any organ system or tissue, can affect more than one body system simultaneously, and can occur at any time after starting treatment or after discontinuation of treatment. Important immune-mediated adverse reactions listed here may not include all possible severe and fatal immune-mediated adverse reactions.
Severe and Fatal Immune-Mediated Adverse Reactions
Immune-Mediated Pneumonitis
Immune-Mediated Colitis
Hepatotoxicity and Immune-Mediated Hepatitis
KEYTRUDA as a Single Agent
Immune-Mediated Endocrinopathies
Adrenal Insufficiency
Hypophysitis
Thyroid Disorders
Type 1 Diabetes Mellitus (DM), Which Can Present With Diabetic Ketoacidosis
Immune-Mediated Nephritis With Renal Dysfunction
Immune-Mediated Dermatologic Adverse Reactions
Other Immune-Mediated Adverse Reactions
Infusion-Related Reactions
Complications of Allogeneic Hematopoietic Stem Cell Transplantation (HSCT)
Increased Mortality in Patients With Multiple Myeloma
Embryofetal Toxicity
Adverse Reactions
Lactation
Hypertension: In differentiated thyroid cancer (DTC), hypertension occurred in 73% of patients on LENVIMA (44% grade 3-4). In advanced renal cell carcinoma (RCC), hypertension occurred in 42% of patients on LENVIMA + everolimus (13% grade 3). Systolic blood pressure ≥160 mmHg occurred in 29% of patients, and 21% had diastolic blood pressure ≥100 mmHg. In unresectable hepatocellular carcinoma (HCC), hypertension occurred in 45% of LENVIMA-treated patients (24% grade 3). Grade 4 hypertension was not reported in HCC.
Hypertension
Cardiac Dysfunction
Arterial Thromboembolic Events
Hepatotoxicity
Renal Failure or Impairment
Proteinuria
Diarrhea
Fistula Formation and Gastrointestinal Perforation
QT Interval Prolongation
Hypocalcemia
Reversible Posterior Leukoencephalopathy Syndrome (RPLS)
Hemorrhagic Events
Impairment of Thyroid Stimulating Hormone Suppression/Thyroid Dysfunction
Impaired Wound Healing
Osteonecrosis of the Jaw (ONJ)
Embryo-Fetal Toxicity
Adverse Reactions
Use in Specific Populations